A study published in Genome Biology systematically compares a panel of BEs for the first time and BEable-GPS is the first database to profile the accessibility of BEs to target pathogenic SNVs. BEable-GPS and its embedded toolsets will provide researchers a great resource for their experimental designs to model or correct disease-related mutations.
This study results from a collaboration between two teams: one led by Dr. Chen Jia from ShanghaiTech’s School of Life Science and Technology and another led by Dr. Yang Li from CAS Key Laboratory of Computational Biology, Shanghai Institute of Nutrition and Health of Chinese Academy of Sciences (CAS). They systematically compare five representative BEs, including BE3, eBE-S3, BE4max, hA3A-eBE-Y130F and dCpf1-eBE for their editing efficiency and product purity at human pathogenic C-to-T sites. The results show that BE4max and hA3A-eBE-Y130F induce higher levels of editing efficiencies than other examined BEs and meanwhile, dCpf1-eBE induces lowest levels of indels than other examined BEs.
Cytosine Base editors (CBE), which combine APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like)/AID with CRISPR/Cas proteins, have been developed to achieve programmable C-to-T changes in one-nucleotide level. Distinct to CRISPR/Cas system, BEs induce efficient base editing in target genomic sites without generation of DNA double-strand breaks (DSBs), which hold great potential to correct and create pathogenic single nucleotide variants (SNVs) for broad application in biomedical studies.
Since the first series of reported BEs, a number of new types of BEs with different Cas proteins, e.g., SpCas9 or dLbCpf1 and various deaminases, e.g. rat APOBEC1 (rA1) or human APOBEC3A (hA3A), have been developed to expand base editing scopes. However, these BEs haven’t been directly compared for their utility in creating or correcting pathogenic point mutations. More importantly, a database comprehensively cataloging pathogenic point mutations that can be corrected or created by different BEs has been lacking.
This incorporated study further profiles the accessibilities of twenty BEs to all reported human pathogenic-related T-to-C or C-to-T point mutations in silico, which revealed that about 94.34% of 17,077 pathogenic SNVs could be generated by at least one BE and 94.28% of 5,031 pathogenic T-to-C SNVs could be corrected by at least one BE. The authors further build a BEable-GPS (Base Editable prediction of Global Pathogenic SNVs) database (website: http://www.picb.ac.cn/rnomics/BEable-GPS) to profile the BE editable pathogenic SNVs and provide gRNA designs for pathogenic SNVs and genomic sites with its embedded toolsets (Fig.1).
This study was supported by grants from National Natural Science of China, Ministry of Science and Technology, Chinese Academy of Sciences and ShanghaiTech University.
Read more at:https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1839-4
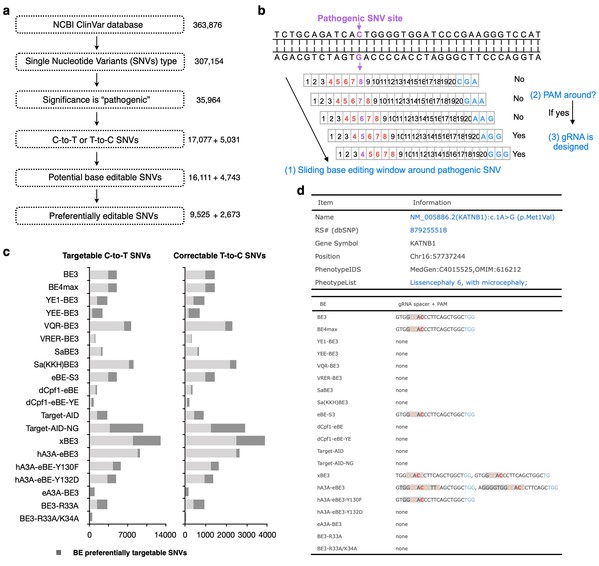
Fig.1 In silico base editable landscape of pathogenic SNVs.