近日,上海科技大学iHuman研究所水雯箐课题组和中国科学院上海有机化学研究所生物与化学交叉研究中心张耀阳课题组合作,在Nature Communications发表了题为“Benchmarking commonly used software suites and analysis workflows for DIA proteomics and phosphoproteomics”的研究论文,通过创建多套标准与复杂样本数据集,全面评估了4种具有不同算法设计的数据非依赖性采集(Data-independent acquisition, DIA) 数据分析软件在解析常规蛋白质组和磷酸化蛋白质组数据时的性能和特点,明确提出了适用于特定类型蛋白质组学的DIA数据分析流程。
基于质谱的蛋白质组学已成为解析生物体系的蛋白质群体组成与动态调控的主流技术。DIA是一种在理论上对样品信息损失极低的质谱数据采集方法,该模式将每个采集循环中一级质量分析器检测的离子分配到预定质量窗口,从而对数个或数十个待测离子同时进行碎裂和二级检测。这种方法保证了数据完整性,但在产出数据的解析上也更加复杂,待测物的碎片离子信号易受干扰,从而会影响数据解析的深度和定量准确性。同时,DIA数据分析通常需要建立参考谱图库与实际数据匹配,不同来源与设计的谱图库也会对最终结果产生很大影响。目前已有多种软件被开发出来用于DIA数据分析,然而对每种软件结合不同谱图库的实际表现缺乏客观和系统性的评价,会在一定程度上影响数据解析的效率、准确性与可重复性。
研究团队首先创建了代表常规蛋白质组学实验复杂性的标准数据集,利用该数据集评估了4种常用软件结合7种谱图库共10条数据分析流程的主要性能(图1),包括蛋白质组鉴定深度、参考谱图库质量对鉴定错误率和打分稳定性的影响、蛋白质组定量准确性和重复性,以及差异蛋白筛选的可靠性。然后利用一套合成磷酸化肽段的标注数据集评估了不同软件对磷酸化修饰位点定位的准确性,并提出了适用于不同软件算法的修饰位点定位打分阈值。最后通过建立实际样本的磷酸化蛋白质组数据集,比较了不同软件解析该数据的能力,揭示了不同数据分析流程在信号通路调控分析上的差异表现(图2)。
对于一个正在快速发展的研究领域,基于DIA方法采集的数据在国际上各实验室不断产出,相应的数据解析算法也不断被开发和升级。该研究对常用DIA数据分析流程的系统性评价,一方面为软件算法开发者提供了高质量测试数据集和对已有方法性能的评估信息,另一方面为DIA组学研究工作者选择合适工具、建立可靠的数据分析方法提供了一份实用的操作指南。
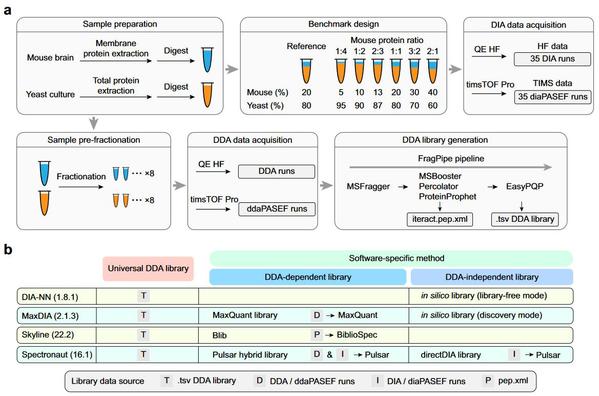
图1.该工作建立的标准DIA蛋白质组数据集以及评价的软件工具与谱图库组合
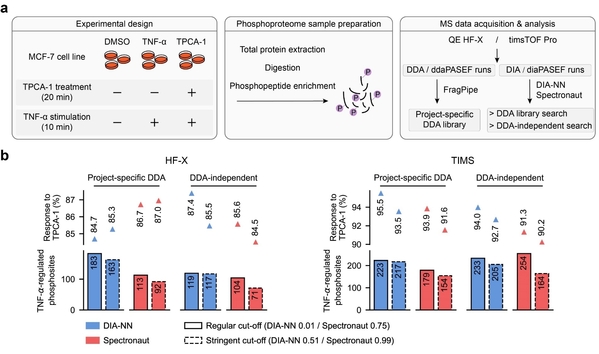
图2. 该工作评价的两种主流软件结合不同谱图库对两套实际样本磷酸化蛋白质组学数据集的部分解析结果
生命学院2020级博士研究生娄容珲、中科院生化交叉研究中心研究生曹野、iHuman研究所研究助理李珊珊为共同第一作者,生命学院2022级博士研究生郎晓雨和中科院生化交叉研究中心李云霞参与了本课题研究。上海科技大学iHuman研究所研究员、生命学院常任副教授水雯箐和中国科学院上海有机化学研究所生化交叉研究中心研究员张耀阳为该工作的共同通讯作者。上海科技大学为第一完成单位。
文章链接:https://www.nature.com/articles/s41467-022-35740-1
 沪公网安备 31011502006855号
沪公网安备 31011502006855号